Overview - Cardiovascular Disease
Cardiovascular disease is the number one cause of death worldwide. Heart failure affects more than 6 million people in the US alone.
Our lab is driven by the goal to understand the molecular mechanisms of these conditions, to help develop novel therapeutic strategies to treat them.
While heart failure is complex, at its core it results in a reduced ability of the heart to fill with blood and then pump it throughout the body. This pumping function is driven by the ability of millions of heart cells (cardio-myocytes) to contract with every heart beat. Inside each of these cells is a specialized protein lattice called the myofilament made up of individual sarcomeres - the molecular motor responsible for the contraction of the heart.
The Cardiac Myofilament - The Engine of the Heart
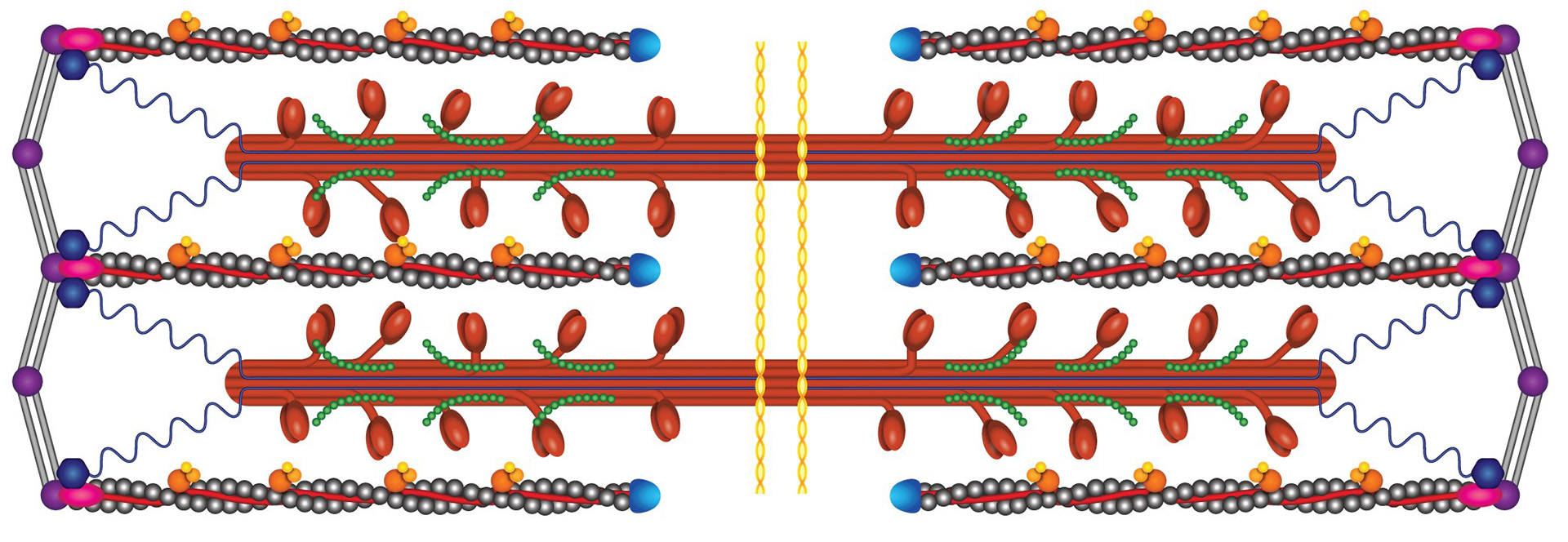
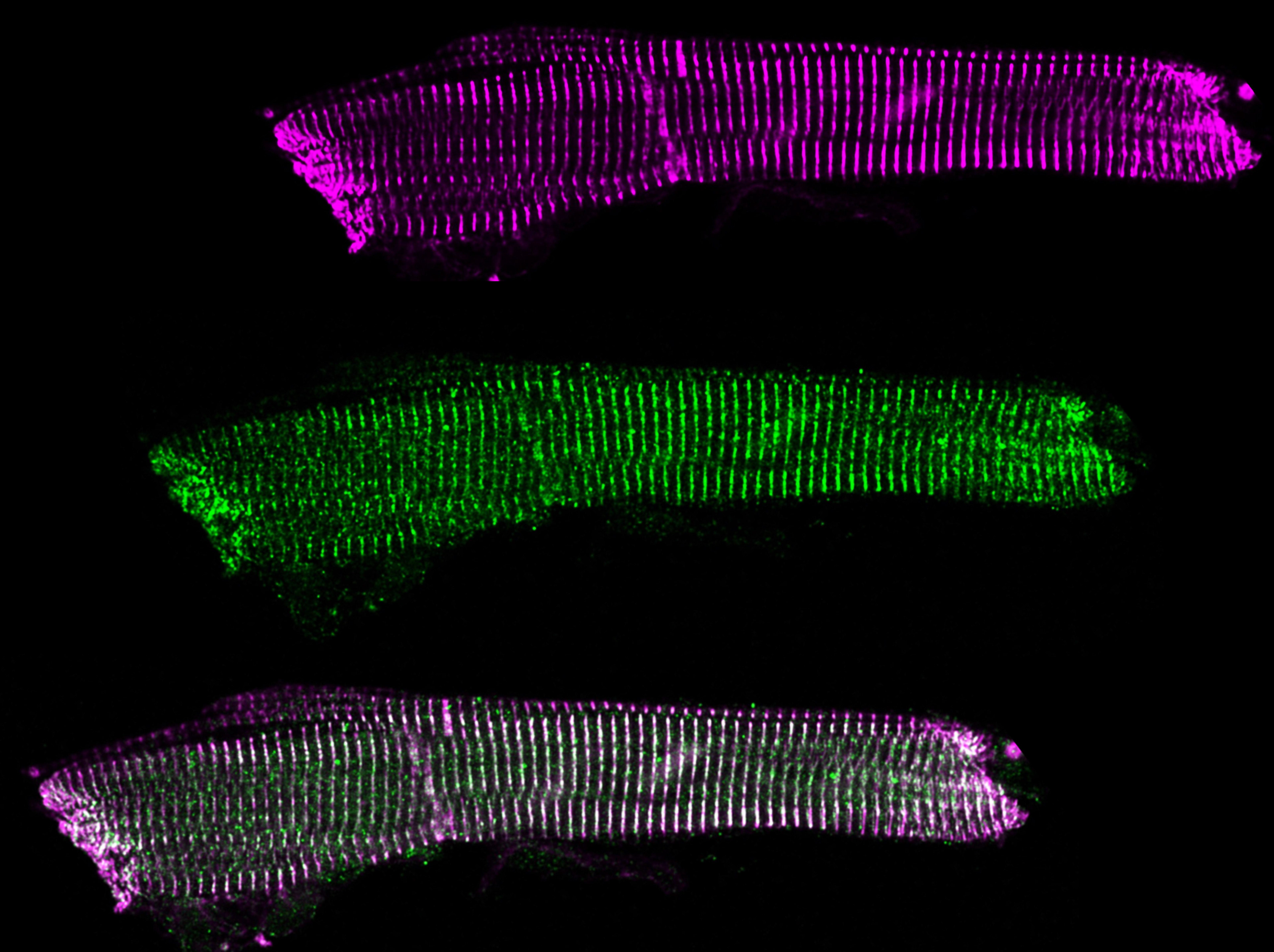
The myofilament is organized into individual sarcomeres.
Within the sarcomere, the thick filament (red in the schematic) is composed of myosin, with myosin heads extending towards the actin thin filaments (grey). When the myosin heads attach to their binding sites on the actin thin filament, they can perform a POWERSTROKE which pulls the filaments past each other, generating force.
When the heart is relaxed and filling with blood (if the heart can’t relax to fill with blood, it doesn’t matter how strong it is), the myosin binding sites on actin are blocked by a protein called tropomyosin that wraps around actin. The troponin complex is the “switch” that moves tropomyosin out of myosin’s binding spot when the heart needs to contract. The signal for troponin to move tropomyosin is intracellular calcium ions that signal the beginning of cardiac contraction.
Proteomics - Peering into the Cell

The myofilament is regulated by protein phosphorylation and other post-translational modifications (PTMs). While antibody-based approaches are useful (and utilized in our lab), they come with a variety of drawbacks including a bias - they can only discover changes that have been looked for.
Mass spectrometry provides an un-biased snapshot at a huge number of proteins and PTMs in a sample. Combined with sample fractionation and enrichment, this allows us to observe even very rare events that can have very significant effects in the heart.
Protein Quality Control and BAG3

The failing heart is associated with a loss of the sarcomere's force generating capacity. While this weakening has been observed for over a half century, the causes are still unknown, hampering our ability to restore contractility in patients.
Our work has shown that a major cause of the contractile malfunction is a loss of normal protein quality control. Indeed, how do you replace the proteins of the sarcomere when the heart cannot stop beating? It would be like replacing your car engine but you cannot stop driving. So even in the healthy heart, this would be a complex challenge for the cell. It is not surprising that the stressed heart would be unable to manage the well orchestrated steps necessary.
A major nexus protein involved in this pathway is BAG3. Through the use of human tissue, animal models, genetic manipulation and gene therapy, super-resolution live cell imaging, cutting-edge mass spectrometry approaches, single cell functional studies, and partnerships with industry, the Kirk lab is actively investigating the foundational science and clinical potential of BAG3 and sarcomere protein quality control pathways.
Mechano-Transduction and GSK-3β
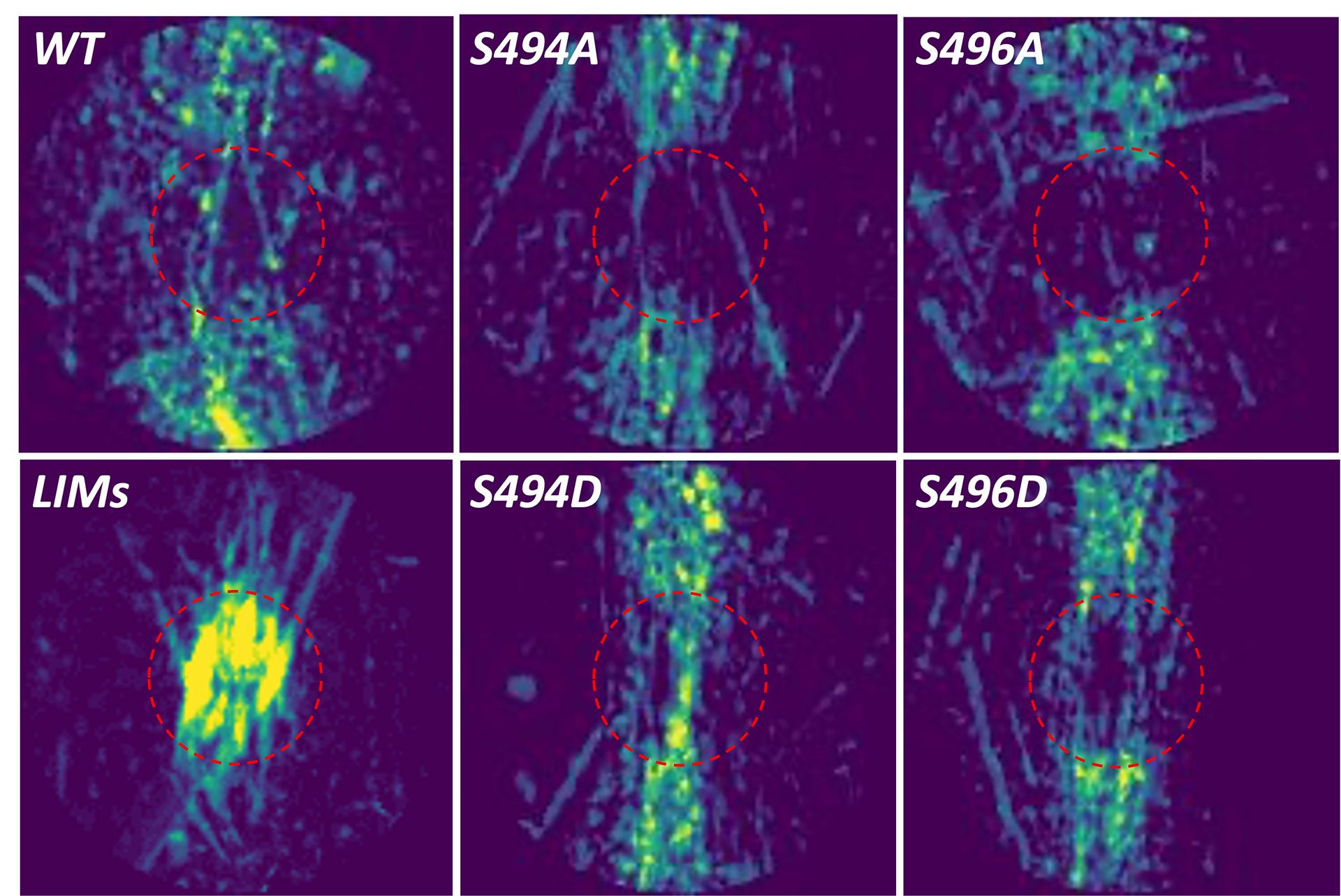
Thirteen decades ago, Otto Frank discovered that when cardiac muscle is stretched, it generates more force. This fundamental mechano-sensing characteristic would become known as the Frank Starling Law of the Heart. It is essential for the heart's ability to match cardiac output to venous return and rapidly respond to changes within the body and the surrounding environment.
The Frank Starling mechanism no longer works in patients with heart failure - worsening lung edema, reducing blood flow, putting additional stress on the right ventricle, decreasing exercise tolerance, and more. Unfortunately, why heart failure patients lose their Frank Starling was known not.
We recently discovered that the kinase GSK-3β localizes to the sarcomere and regulates Frank Starling. Unfortunately, patients with heart failure have significantly reduced GSK-3β activity, leading to the loss of Frank Starling. The Kirk lab's ongoing work aims to understand this fundamental law of physiology and how to restore it in heart failure patients, using novel genetic animal models, advanced laser ablation studies, sophisticated biophysical assays, small molecule treatments, high-energy x-ray diffraction, and other techniques.
Methylglyoxal: A Recipe for Cardiovascular Complications
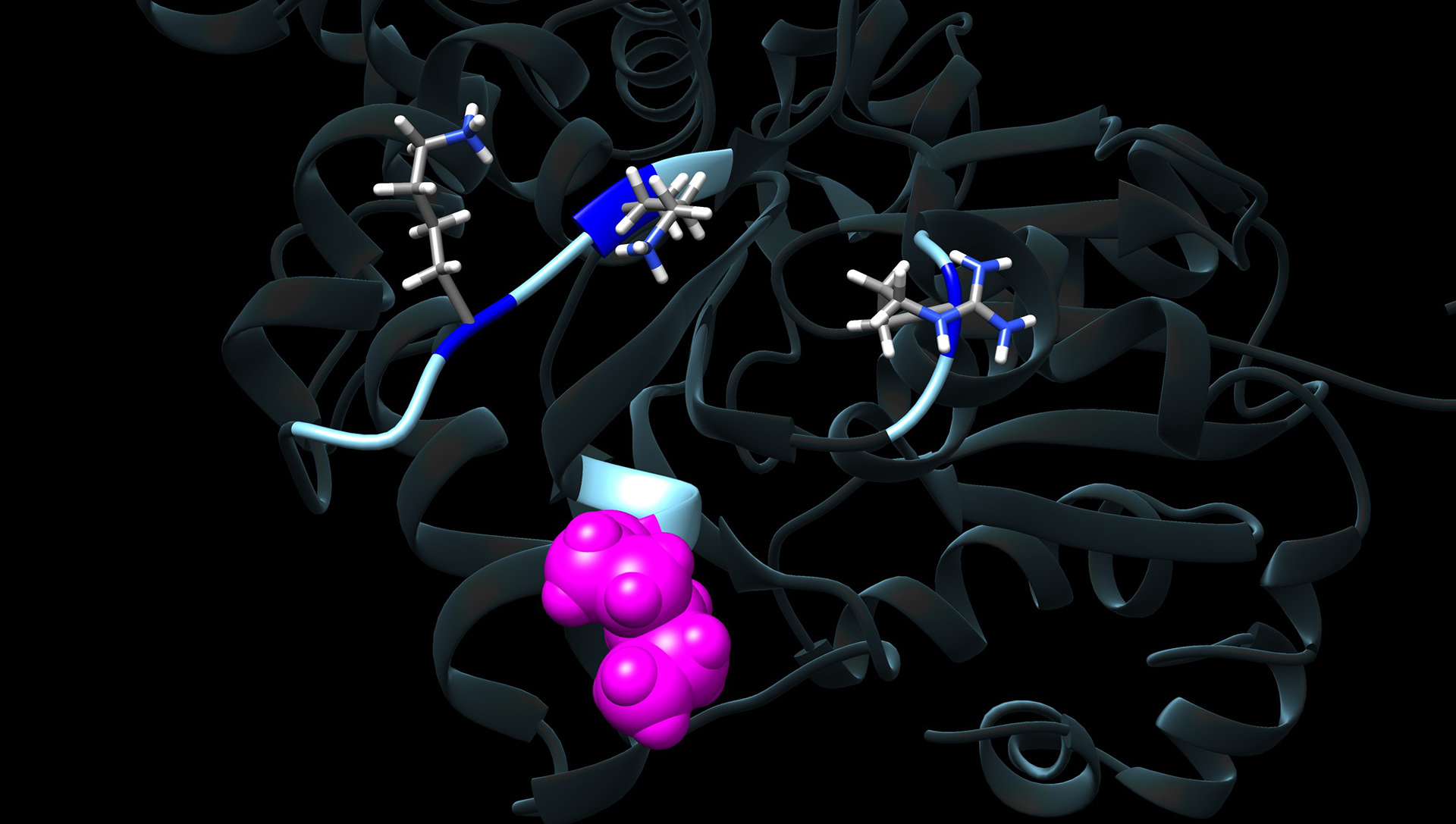
There are multiple conditions that significantly increase the chances of developing heart failure, such as obesity, diabetes, smoking, and aging. The molecular mechanisms by which these increase heart failure risk are not known, but they all lead to an increase in a small reactive carbonyl species known as Methylglyoxal (MGO).
MGO can irreversibly modify intracellular proteins, and we have shown that many of the risk factors described above increase MGO in the cardiomyocyte. This MGO interacts with sarcomere proteins and dramatically interferes with their ability to generate force.
Unfortunately, MGO modifications are irreversible, and past attempts at reducing MGO clinically have failed. Our new approach is to discover how MGO is inducing dysfunction - and correct that dysfunction at the molecular level. These efforts involve clinical studies, mouse models of aging and metabolic disorders, computer simulations, structural analyses, single molecule approaches, mass spectrometry and more.